



Introduction
Amyloidosis refers to a group of disorders in which usually soluble proteins become insoluble, fibrous proteins, referred to as amyloids. In humans, there are 25 different proteins that can take up this fibrillar conformation in vivo (Gillmore and Hawkins, 2013). Amyloids are deposited in the extracellular space of almost any organ or tissue (Murakami et al., 2014). Extracellular amyloid deposits are associated with a breakdown in organ function and lead to the development of clinical syndromes, whose categorization depends on the causative fibril protein precursor.
Amyloids form due to a disorder in the secondary structure of proteins which causes the protein to assume an aggregative, β-pleated sheet conformation (Elsevier’s Global Clinical Reference, 2012). If amyloid accumulation occurs in a single organ, amyloidosis is localized. In systemic amyloidosis, amyloid deposits are widespread throughout the body and usually accumulate in a progressive manner (Gillmore and Hawkins, 2013). All amyloid deposits contain pentraxin serum amyloid P (SAP) and glycosaminoglycans.
At least 24 such proteins have been recognized as causative of amyloidosis. Diagnosis depends on the identification of amyloid deposits in biopsy or autopsy tissue. Treatment aims at reducing the production and extracellular deposition of amyloids, promoting the lysis of existing amyloids, as well as treating dysfunction of the underlying organ. Systemic amyloidosis is usually a progressive disease. If untreated, 80% of patients die within 2 years of diagnosis (Elsevier’s Global Clinical Reference, 2012).
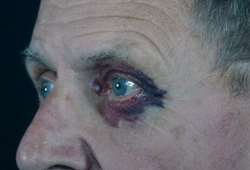
Figure 1. Amyloidosis-associated periorbital peliosis ( Gertz, 2013 )
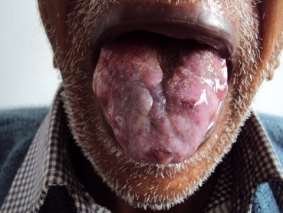
Figure 2. The massive expansion of the lingua caused by starchlike infiltration ( Mayankaet Al., 2010 )
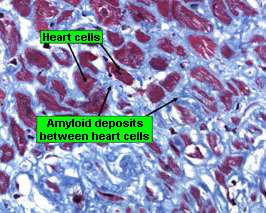
Figure 3. Amyloid fibrils discoloration blue. Amyloid sedimentations are seen everyplace and encircle each bosom cell ( Stanford Hospital & A; Clinics, 2014 )
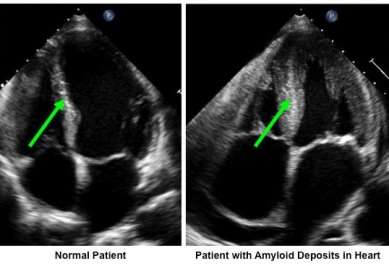
Figure 4. Heart ultrasound from a normal patient and one with amyloid sedimentations in the bosom. The thickness in the patient with amyloidosis is not due to excess bosom muscle, but instead from sedimentations of amyloid filaments (Stanford Hospital & Clinics, 2014).
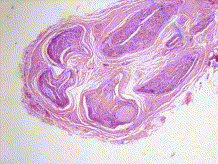
Figure 5. Sural nerve biopsy for amyloid involvement of peripheral nervous system (PNS). The nodular sedimentation of pink formless material in the subepidermal layer is the amyloid (Gertz, 2013).
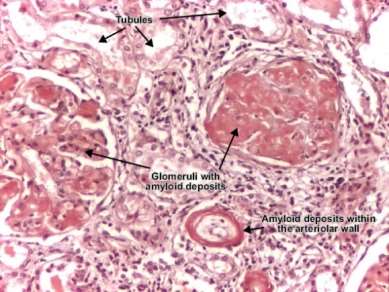
Figure 6. Amyloid deposits in the kidney (Gertz, 2013).
Systemic Amyloidosis – Subtypes and Clinical Features
Systemic amyloidosis is divided into three major subtypes depending on the causative protein:
A. Primary amyloidosis (AL) occurs when the bone marrow produces excessive amounts of Igs by plasma cells that build up in the bloodstream and deposit in tissues. It accounts for more than 80% of all cases. The causative protein is immunoglobulin light chains. The clinical characteristics of primary amyloidosis are variable, as it may affect any organ other than the CNS (Gillmore and Hawkins, 2013). This form is associated with an underlying plasma-cell dyscrasia (a group of diseases identified by the proliferation of a single clone of cells producing a monoclonal Ig or immunoglobulin fragment: multiple myeloma, Waldenstrom macroglobulinemia, the heavy-chain disease, benign monoclonal gammopathy).
AL consistently affects the kidneys, heart, and peripheral nervous system (PNS). More than 60% of affected persons present with renal dysfunction, which causes proteinuric renal failure. Half of the affected persons also develop stage 1 or 2 chronic kidney disease (CKD), while 16% develop stage 5 CKD (Gillmore and Hawkins, 2013). This form exhibits the broadest spectrum of organ involvement. AL may lead to carpal tunnel syndrome, cardiomyopathy and congestive heart failure, enteric malabsorption, liver swelling, kidney failure, nephrotic syndrome, neuropathy, and orthostatic hypotension.
Symptoms depend on the organs in which amyloids have accumulated. They include abnormal heart rhythm, swollen tongue, fatigue, numbness of hands or feet, shortness of breath, skin changes, swallowing problems, swelling of limbs, and weight loss (Gertz, 2011). Treatment aims to reduce plasma cell dyscrasia. It involves high-dose Alkeran chemotherapy together with Decadron, then peripheral stem cell transplantation. Additional measures are taken to treat dysfunction of the underlying damaged organ (Elsevier’s Global Clinical Reference, 2012).
B. Familial amyloidosis (ATTR). Familial amyloidoses are autosomal dominant inherited diseases. The amyloid is synthesized from birth, but deposits occur mid-life (Amyloidosis Foundation, 2011). Familial amyloidoses include amyloidoses ALys, AApoA1, AFib, and amyloidosis associated with apolipoprotein AA¬ II- (AApoA2) (Gillmore and Hawkins, 2013), with ATTR being the most common. ATTR accounts for about 5% of systemic amyloidosis cases. It’s caused by familial mutations of particular protein groups that form filaments, typically transthyretin (TTR), which is produced in the liver (Elsevier’s Global Clinical Reference, 2012). Renal dysfunction in familial amyloidoses is common, while ATTR predominantly affects the heart and less often the PNS, causing megalocardia and peripheral neuropathy.”
Symptoms of ATTR are common in AL
For this type of amyloidosis, there is no specific pharmacotherapy available, merely supportive therapy. In instances of extended liver engagement, a liver organ transplant is required (Elsevier’s Global Clinical Reference, 2012) to take the portion of the tissue where mutation TTR is produced and replace it with healthy tissue with normal TTR production. This prevents the patterned advance of autonomic and peripheral neuropathy but exacerbates cardiomyopathy. Patients with extensively affected kidneys may also benefit from following a dietary regimen with limited protein, Na, K, and P (Amyloidosis Foundation, 2011).
C. Secondary amyloidosis (AA). A form that develops simultaneously with a chronic infectious or inflammatory disease, for example, TB, rheumatoid arthritis, osteomyelitis, etc. It accounts for approximately 5% of all cases. The causative protein is serum amyloid A protein (SAA). Approximately 97% of patients with AA present with proteinuric kidney dysfunction, and more than 50% have nephrotic syndrome. Finally, 40% progress to end-stage renal disease (ESRD). AA amyloids systematically infiltrate the spleen, and in 33% of cases, the adrenal glands. This results in hepatosplenomegaly in 9% of individuals (Gillmore and Hawkins, 2013).
The heart is not usually affected (only 10% of cases) (Elsevier’s Global Clinical Reference, 2012). Patients with extensively affected kidneys may also benefit from following a dietary regimen with limited protein, Na, K, and P (Amyloidosis Foundation, 2011). Symptoms include bleeding in the skin, fatigue, irregular pulse, numbness of extremities, rash, shortness of breath, swallowing troubles, swollen arms or legs, swollen tongue, and weight loss. Treatment involves treating the coexisting inflammatory disease as well as the dysfunctional organs. A renal organ transplant may be a necessity (Elsevier’s Global Clinical Reference, 2012).
Aetiology and Pathogenesis – Molecular Basis of Systemic Amyloidosis
A change in a protein’s secondary structure results in a misfolded protein, the amyloid. Amyloids tend to cluster with other amyloids to form filaments, which accumulate in the interstitium and distort organ function. Factors contributing to this tendency include:
- Protein Concentration is pathologically increased, e.g., serum amyloid A protein in chronic inflammatory diseases in ESRD.
- A protein that has an inherent tendency for misfolding and clustering, e.g., familial proteins in familial amyloidoses.
- A protein that is proteolytically remodeled, e.g., cleavage of inherent memA¬brane protein 2B by furin peptidase and release of amyloid A¬? peptides by secretases.
- Age, e.g., wild-type TTR is amyloidogenic and is associated with age-related amyloid deposition.
The occurrence, timing, extent, and effects of starch-like accretion are not merely dependent on these factors but also on environmental and genetic constituents. For example, the amyloidogenic Val30Met discrepancy of TTR exhibits variable forms in infiltration and clinical characteristics among different cultural groups (Gillmore and Hawkins, 2013).
Amyloid structure
Electron microscopy and X-ray diffraction analyses have shown that amyloid sedimentations are composed of stiff, non-branching filaments with an average diameter of 7.5-10 nanometers and a cross-β-sheet super secondary construction (Gillmore and Hawkins, 2013).
Components of amyloid sedimentations
Serum amyloid P (SAP) constituent – a circulating glycoprotein of the pentraxin family – is bound by all types of filaments through a particular Ca2+-dependent interaction which fractionally protects the filaments from proteolysis. Amyloid deposits also typically contain numerous proteoglycans. For example, heparan sulfate was shown to be involved in the formation of amyloid sedimentations since degradation of heparan sulfate by heparanase inhibits the initiation of AA. Heparan sulfate hastens the transition of serum amyloid A (SAA) from its native into the amyloidogenic conformation and also accelerates the formation of filaments by amyloidogenic SAA, TTR, Ig light chains, and amyloid-β through selective binding to a basic motive. Amyloid deposits also include laminin and type IV collagen (ECM constituents) as well as chaperone proteins such as apolipoprotein E and clusterin (Gillmore and Hawkins, 2013).
Dynamics of fibril formation.
Studies in vitro demonstrated that the formation of amyloid filaments proceeds via nucleated growth similar to crystallization. Initially, a slowdown stage occurs in monomeric proteins. A critical karyon is generated, and fibril formation begins and proceeds very fast. Amyloidogenic proteins with a conformation prone to the collection are incorporated into the growing filaments. The rate of filament formation is much faster than the natural clearance of amyloid.
Amyloid traces in tissues may remain even after an optimum response to therapy. In the case of disease relapse, these residues can quickly reconstruct amyloid sedimentations. These in vitro findings are relevant in cases of AA in which control of SAA production and progressive decrease in albuminuria if followed by a short increase in SAA levels, result in deteriorated renal function (Gillmore and Hawkins, 2013).
Organ tropism. Certain misfolded proteins have a tendency for deposition in certain organs. One example is leukocyte chemotactic factor 2 (LECT2) deposits in the kidney in AA. Another one is TTR’s preference for peripheral nerves in ATTR. The exact mechanism of organ tropism is unclear, though factors that contribute may include cellular receptors, pH, local protein concentrations, tissue-specific glycosaminoglycans, interactions with collagen, or specific local proteolytic enzymes (Gillmore and Hawkins, 2013).
Mechanisms of tissue damage. The precise mechanism of tissue damage by amyloid sedimentations is not fully understood. Some evidence suggests possible cytotoxicity of pre-fibrillar oligomeric species, such as TTR, Ig light chains, amyloid-β protein, and prions. Clinical evidence of such cytotoxicity was best observed in cardiac AL;
After chemotherapy-induced suppression of amyloidogenic light chains, the serum concentration of a marker of cardiac dysfunction, N-terminal pro-brain natriuretic peptide (NT-proBNP), may well and quickly decrease (Gillmore and Hawkins, 2013).
Epidemiology
Systemic amyloidosis is responsible for about 1 in 1,500 deaths yearly in the UK and apparently also in other developed countries. Amyloidoses mainly manifest in mid to late life, but typically AA can also occur in children. About 4% of adult renal biopsies reveal nephritic amyloidosis. The disease develops in ~2% of individuals with monoclonal B-cell dyscrasias. AA amyloidosis may complicate about any chronic inflammatory condition, and the reported prevalence in patients with chronic arthritides is 3–6%. The incidence of AA is higher in Europe than the USA. Worldwide, AA incidence is gravitating for unknown reasons. Between onset of inflammation and diagnosis of AA, the duration of latency exhibits a median of 17 years. In the UK, the prevalence of familial non-neuropathic amyloidosis, including AFib, apolipoprotein AI-associated and lysozyme-associated (ALys) amyloidoses, is about 1.5 instances per million (Gillmore and Hawkins, 2013).
( Survival )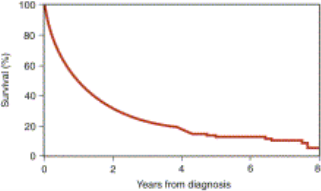
( Old ages from Diagnosis )
Figure 7. Overall endurance was seen in MayoClinic of patients with Amyloidosis ( Gertz, 2013 )
Treatment
Systemic AL amyloidosis. AL treatment depends on age, cardiac function, and treatment toxicities. Currently, the universally applied treatment regimens for AL include the following combinations:
- bortezomib, cyclophosphamide, and dexamethasone,
- melphalan and dexamethasone,
- cyclophosphamide, thalidomide, and dexamethasone, and
- lenalidomide and dexamethasone (Gillmore and Hawkins, 2013).
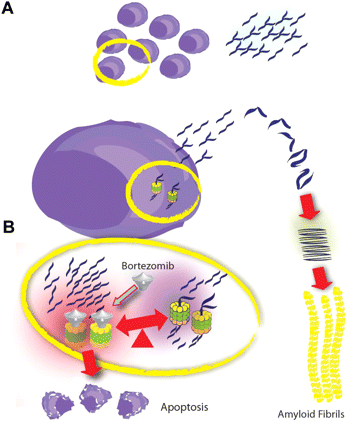
Figure 8. A small plasma cell ring produces amyloidogenic light chains which misfold and form amyloid filaments (A). Inside the plasma cell, increased protein burden induces ER stress and rapid elimination of these proteins is required to maintain homeostasis. The degradation of these proteins depends on proteasome activity. Bortezomib blocks proteasome degradation of proteins and increases poor quality protein burden within the ER, inducing ER stress beyond the capacity of the control mechanisms and resulting in plasma cell apoptosis (B) (Dimopoulos & Kastritis, 2011).
AA amyloidosis. Treatment aims to suppress the underlying inflammatory disease to the highest possible degree by reducing the production of SAA. SAA concentration is a strong predictor of survival and renal outcome. Complete and continuously suppressed inflammation achieved together with the establishment of normal SAA levels (<4 mg/l) correlates with an 18-fold lower mortality risk compared to SAA levels ?155 mg/l. About 40% of patients with AA eventually require renal replacement therapy, with a median time to dialysis from diagnosis of about 6 years (Gillmore and Hawkins, 2013).
Familial amyloidosis. Familial non-neuropathic amyloidosis tends to be “silent” for several decades. In apolipoprotein AA-I- A-associated and lysozyme-A-associated types, kidney transplantation for ESRD is usually highly successful, and transplants are functional for decades. In the case of AFib, amyloid deposits reappear and lead to graft loss after about 7 years. Another possible path is combined hepatorenal transplantation since factor I is synthesized solely in the liver, but this option poses an increased mortality risk. ATTR is generally fatal within 5-15 years. A placebo-controlled trial in 125 patients with early-stage ATTR (Val30Met variant of TTR) suggested that tafamidis, a small molecule that converts circulating TTR into its stable conformation, therapy might slow disease progression (Gillmore and Hawkins, 2013).
( Survival )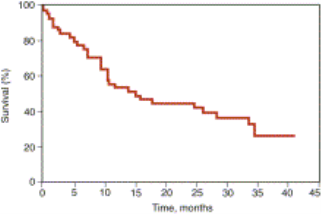
( Time, Months )
Figure 9 shows the survival of 55 patients with amyloidosis treated with high-dose Decadron (Gertz, 2013). Dexamethasone is a glucocorticoid agonist that crosses cell membranes and binds with high affinity to specific cytoplasmic glucocorticoid receptors. The complex binds to DNA elements, leading to alterations in written text and protein synthesis that suppress leukocyte infiltration at the site of inflammation, block mediators of the inflammatory response, and reduce swelling (Drugbank, 2013).
Mentions
- Amyloidosis Foundation Authors. ( 2011 ) . “ Familial ” .
- Dimopoulos, MA. & A ; Kastritis, E. ( 2011 ) “ Bortezomib for AL amyloidosis: traveling frontward ” , Blood, 118, p827-828. Department of the Interior: 10.1182/blood-2011-05-355115.
- Drugbank writers, ( 2013 ) . “ Dexamethasone ” .
- Elsevier’s Global Clinical Reference: ClinicalKey ( 2012 ). “ Amyloidosis ”
- Gertz, MA. ( 2011 ) . “ Amyloidosis ” . Cecil Medicine, chap 194.
- Gertz, MA., Buadi, FK., Zeldenrust, SR., Hayman, SR. ( 2013 ) Hematology: Basic Principles and Practice, 9, p1350-1374
- Gillmore, J. D. & A; Hawkins, P. N. ( 2013 ). “ Pathophysiology and intervention of systemic amyloidosis ”. Nature Reviews Nephrology, 9, p.574–586. doi:10.1038/nrneph.2013.171
- Mayanka, B., Ritu, K., Cherry, B. & A; Jalees, F. ( 2010 ) Egyptian Dermatology Online Journal,6, p.9
- Murakami, T. , Ishiguro, N. & A ; Higuchi, K. ( 2014 ) . “ Transmission of Systemic AA Amyloidosis in Animals ”. Veterinary Pathology, 51, p.363–371. doi:10.1177/0300985813511128
- Stanford Hospital and Clinics, ( 2014 ). “ AL ( Primary ) Amyloidosis ”
